Research

Getting involved with research in evolutionary biology has been a long time dream of mine, and I feel extremely fortunate that I have been gifted so many opportunities to be able to contribute to so many projects during my time as an undergraduate student. I work on projects with two primary groups: the Rosenthal Lab based in the Department of Biology at the University of Padua, and the Blackmon Lab based in the Department of Biology at Texas A&M University. My work with the Rosenthal Lab focuses on morphometrics and computational forward time simulation of early generation hybrid populations in poecilid fish (Xiphophorus birchmanni and X. malinche), focusing particularly on the interactions between polygenic traits that are targets of mate choice and ecological selection. My work in the Blackmon Lab encompasses a variety of projects studying the evolution of genome structure, with a special emphasis on sex chromosome evolution and the development/application of phylogenetic comparative methods.
Drivers of morphometric differentiation in early genetation hybrid Xiphophorus swordtails
PIs: Drs. Gil Rosenthal and Heath Blackmon
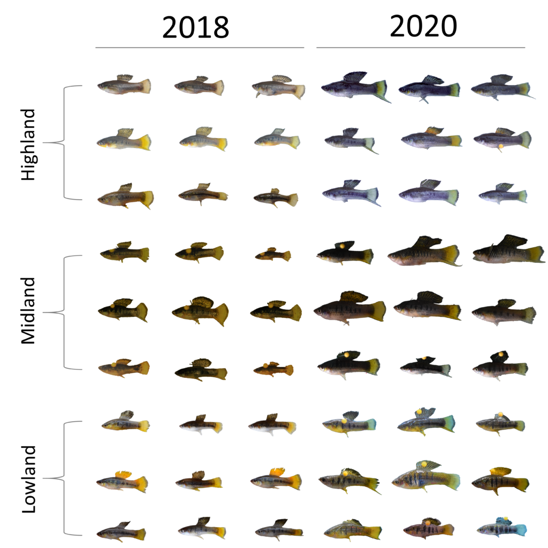
With advances in sequencing methods corresponding with more access than ever to sequence data, it has become increasingly apparent that naturally occurring hybridization events are significantly more common than they were once thought to be. When two hybridizing species are differentiated along multiple axes, hybrids between them are able to explore a wide range of trait space and populations of these hybrids are diverse in phenotype. When diverse phenotypes are combined with diverse selection regimes, populations differentiate rapidly. The components of selection that drive differentiation in populations following gene flow can often be difficult to tease apart using purely empirical approaches, but computational simulations facilitate easy exploration of a wide variety of selection regimes. If both empirical and computational methods are applied in conjunction, statistical model fitting approaches can then be used to infer the selection regime that best corresponds with phenotypes observed empirically.

My work in the Rosenthal Lab applies these concepts to a system revolving around two species of naturally hybridizing Xiphophorus swordtail fish, X. birchmanni and X. malinche. These sister species are diverged morphologically, physiologically, and behaviorally, with differentiated habitat preferences and mate choice behavior resulting in discordance between the selective pressures encountered by each species. Hybrids between the two species exhibit diverse morphology, physiology, and behavior, representing axes along which hybrid populations can differentiate. As part of an ongoing project funded under the NSF’s Long Term Research in Environmental Biology grant, the Rosenthal lab monitored replicated mesocosm hybrid populations at regular intervals over a five year period, collecting a variety of morphometric data from individuals in the populations. To complement this extensive empirical dataset, I designed individual-based forward time population simulations that replicate the structure of our empirical populations. These simulations incorporate up to six polygenic traits which have genomic architecture informed by empirical studies on quantitative trait loci in the system, and implement various combinations of selection functions and strengths, including ecological selection, incompatibility selection, and various forms of mate choice. Where sufficient empirical phenotypic data was available (at this point in time, long term empirical data only exists for morphometric measurements), sum mean error approaches were used to determine the selection regime that resulted in the simulated phenotypes which most closely fit empirical phenotypes.
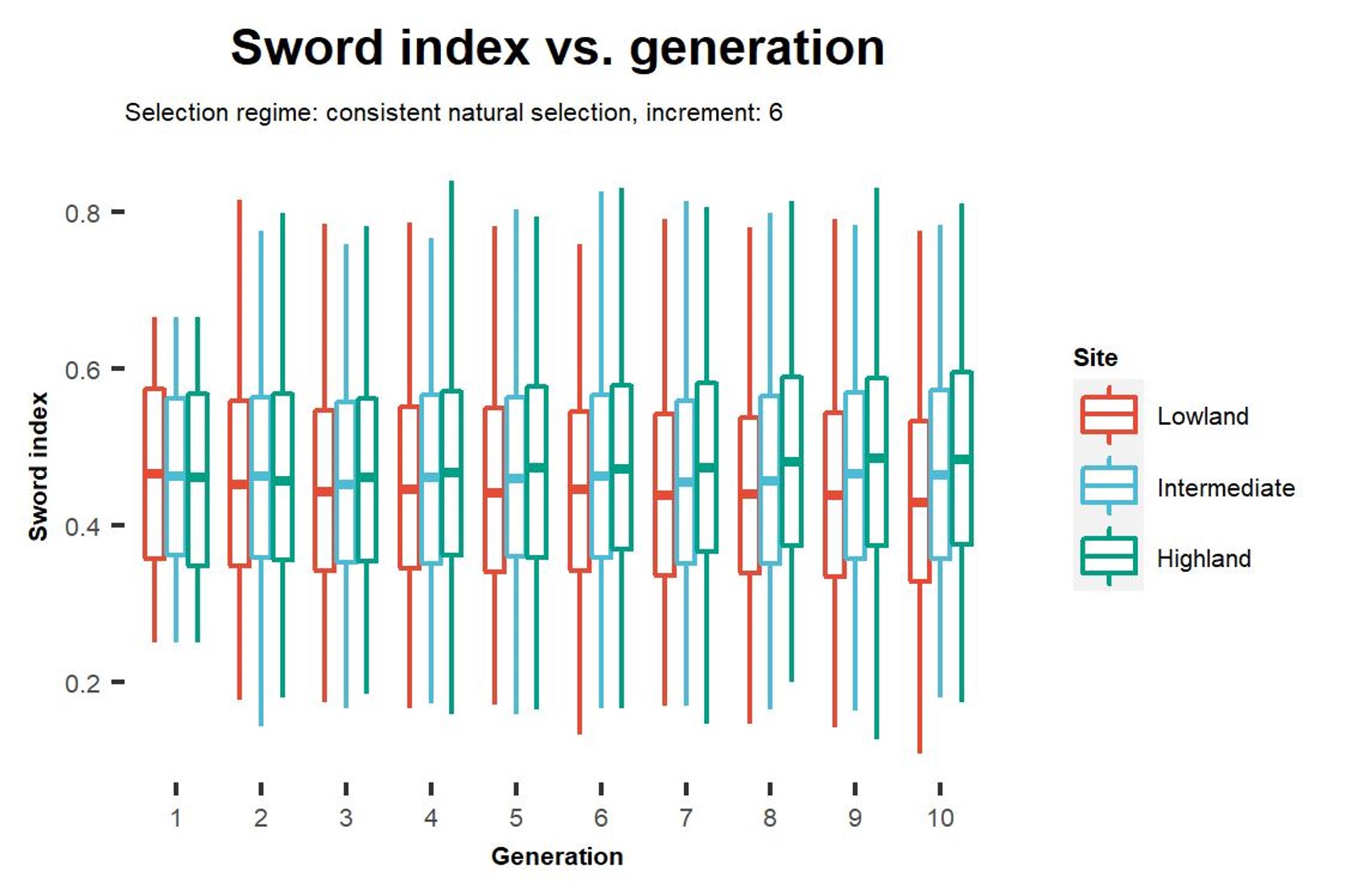
One of the major findings from this project is that direct selection on traits which are polygenic can lead to differentiation in other polygenic traits which are not direct targets of selection. This results from physical linkage between loci which contribute genetic variation to different traits, some degree of which is bound to occur when traits are highly polygenic. It is for this reason that we observe in our simulations that differentiation in sword index, a morphometric trait which is relevant to mate choice, is driven primarily by ecological selection on thermal tolerance, a physiological trait. This "dragging" along of traits has long been hypothesized as one of the primary drivers of morphometric differentiation in our empirical mesocosm populations encountering different environmental conditions, and using simulations we are able to show that this does indeed occur. We demonstrate that in early generation populations encountering multiple sources of selection pressure, differentiation in display traits relevant to mate choice is driven by indirect selection through physical linkage with physiological traits which are under ecological selection. We suggest that direct selection on display traits by means of sexual selection is hindered by the need for resetablishment of linkage disequilibrium between loci associated with display traits and preference for these display traits follwing admixture and the associated breaking of linkage disequilibrium.
Sex chromosome autosome fusions in mammals
PI: Dr. Heath Blackmon
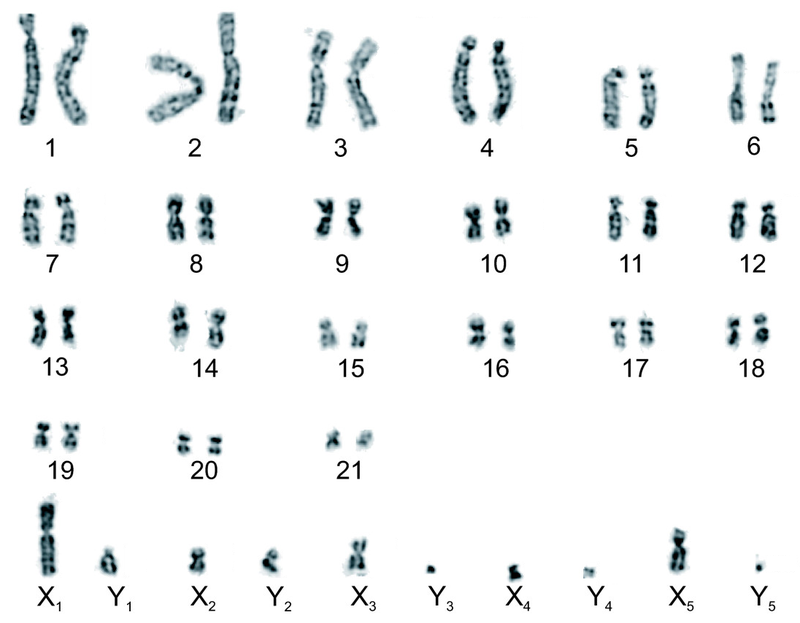
Fusions between chromosomes are a key mechanism by which genome structure evolves. Fusions between autosomes and sex chromosomes are especially interesting due to the potential they have to form new associations between sex and loci which were historically autosomal. There are certain conditions under which the formation of such new associations may have beneficial effects on fitness. One of these conditions is the presence of intra-locus sexual antagonism at an autosomal locus. Intra-locus sexual antagonism describes a situation where an allele at an autosomal locus confers differential fitness effects which are dependent on sex. Because the polarity of fitness effects associated with loci experiencing sexual antagonism is dependent on sex, a possible mechanism towards the resolution of intra-locus sexual antagonism lies in the formation of non-random associations between sex and inheritance of the allele in question. Fusions between sex chromosomes and autosomes have the potential to form these non-random associations, and as such, resolution of intra-locus sexual antagonism has long been hypothesized as a potential selective pressure driving sex chromosome-autosome fusion (SAF) events. However, attempts to find empirical evidence for positive selection on SAF events have been mostly restricted to smaller taxonomic clades, and often return mixed results. The best support for broad selection for SAF events would be an excess of such fusion types on large taxonomic scales. Diverse in karyotype and heterogenous in rates of karyotypic evolution, mammals represent an interesting taxonomic class upon which to evaluate this hypothesis and utilize methods which have previously only been applied to smaller taxonomic clades on a limited scale
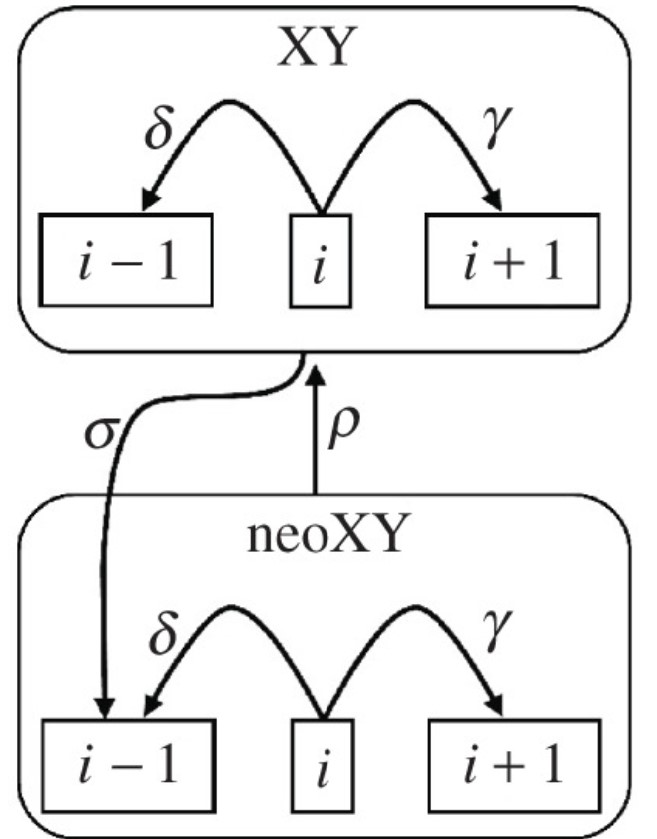
This project incorporates an extensive database of mammalian karyotypes that has been maintined by the Blackmon Lab, a modern timetree of mammalian taxa which incorporates genomic data, and a biologically realistic markov model describing the evolution of karyotype, expanding on phylogenetic comparative methods used in the study of discrete character evolution to explore the evolution of karyotype across time in 950 mammal species. We estimate rates associated with our model of character evolution using Bayesian MCMC methods, and use stochastic mapping techniques to produce maps of ancestral karyotypes and fusion events. We develop new methods to "fill the gaps" associated with stochastic maps when rate heterogenity throughout the phylogeny being mapped is prominent, expanding the applicapbility of this commonly used phylogenetic comparative method. Observed SAF events as a proportion of total fusion events are extracted and compared against proportions expected given a null hypothesis of no selection on fusions of any kind.
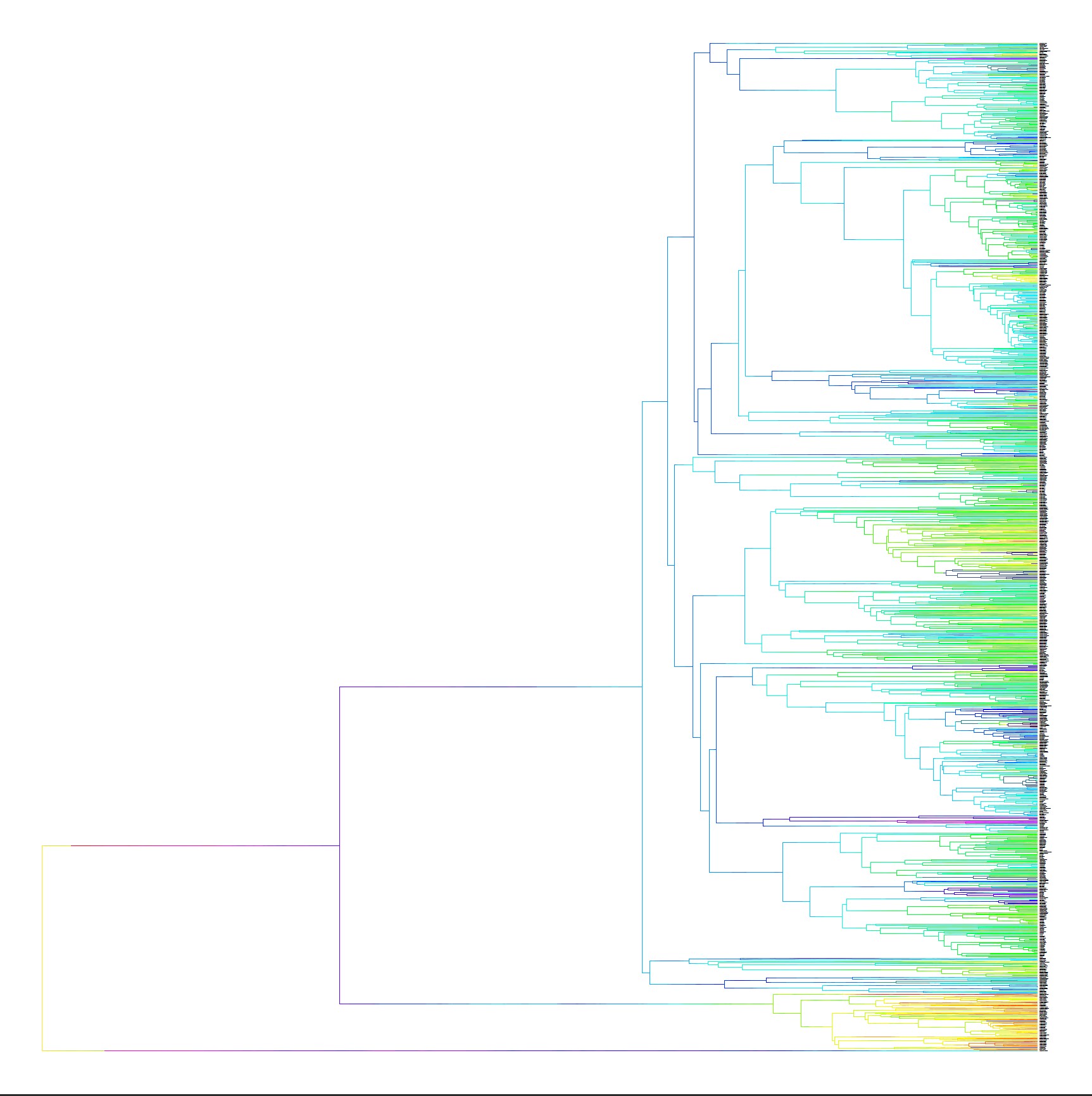
Our preliminary results suggest that observed proportions of SAF events are significantly lower than proportions expected under a null model, with this being true of both mammals as a whole and several mammal subclades. We hypothesize that this is driven by high rates estimated for autosome – autosome fusions and fissions in our model. Various factors such as extreme rate heterogeneity and the inability of our current model to account for multiple SAF events may contribute to this overestimation. Our next steps are to develop a new model which allow for such multiple fusions and rerun the analyses under this new model. We predict that this should result in null and observed SAF proportions that are more aligned.
Developing methods to quantify and visualize heterogenity in discrete character evolution across phylogenies
PI: Dr. Heath Blackmon
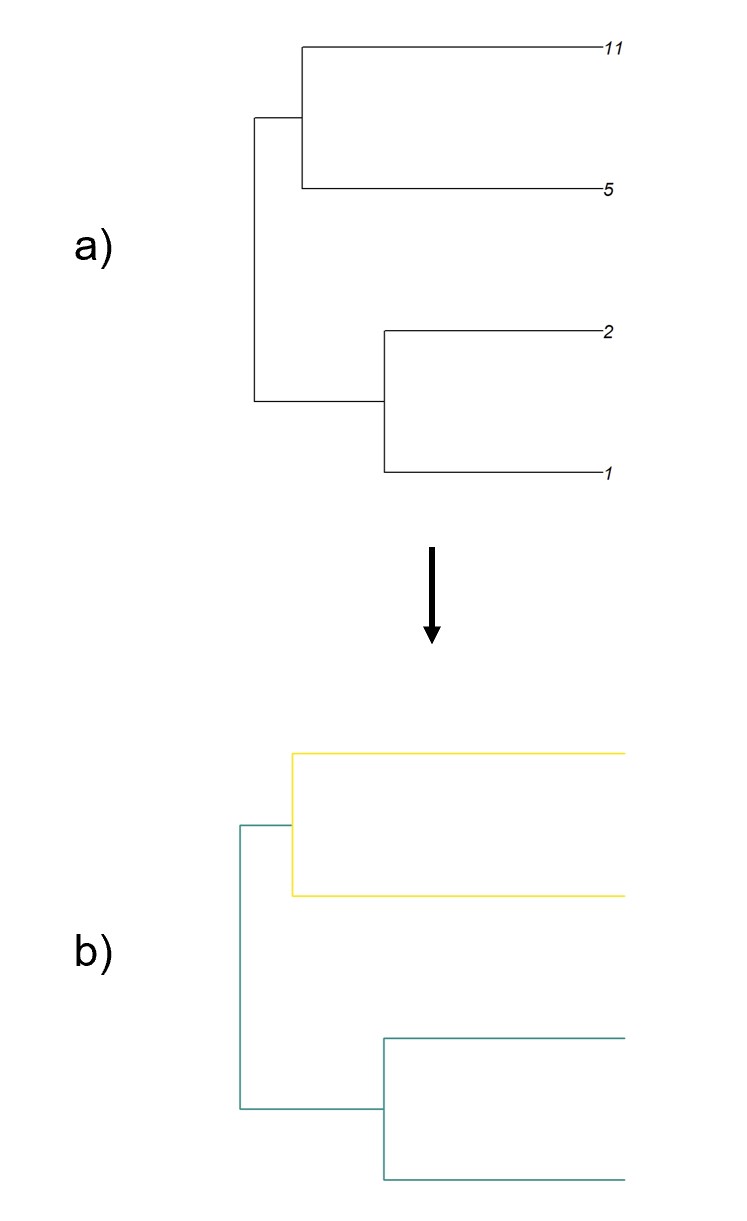
When trying to estimate rates of discrete character evolution, most phylogenetic comparative methods attempt to find a single rate which can be applied to an entire tree. This resulting parameter represents a compromise between rates estimated for portions of the tree where the trait changes rapidly and portions of the tree where the trait remains stagnant. Understanding which portions of the tree push rates up and which portions of the tree push rates down can be insightful in understanding the factors which correlate with rapid rates of change in a trait.
We develop a method to allow for edge-by-edge analysis of rate heterogeneity, built around assigning edges to a set of scalar bins centered around one which are associated with length extensions or reductions. We begin by first taking a user supplied model, phylogeny, tip states, and estimating rates using existing maximum likelihood methods. We assume that the root of the tree has a scalar of one (no change in edge length). We then perform a preorder traversal of the tree and allow for each edge to have possible scalars of either the scalar associated with the parent edge, the scalar one bin above, or the scalar one bin below. At each edge, the lengths of previously traversed edges are multiplied by their assigned scalars, and the length of the current edge is independently multiplied by each of the possible scalars. The likelihood of observing the rates initially estimated given the modified edge lengths is calculated for each potential current scalar, and the potential scalar which maximizes likelihood is assigned to the current edge. This results in each edge of the phylogeny having an associated scalar describing whether the edge has more or less transitions than expected given the length of the edge and estimated rates. We also develop a function that allows for the coloring of edges based on associated scalars. Together, these functions allow for easy quantification and visualization of rate heterogenity in discrete character evolution.
We develop a method to allow for edge-by-edge analysis of rate heterogeneity, built around assigning edges to a set of scalar bins centered around one which are associated with length extensions or reductions. We begin by first taking a user supplied model, phylogeny, tip states, and estimating rates using existing maximum likelihood methods. We assume that the root of the tree has a scalar of one (no change in edge length). We then perform a preorder traversal of the tree and allow for each edge to have possible scalars of either the scalar associated with the parent edge, the scalar one bin above, or the scalar one bin below. At each edge, the lengths of previously traversed edges are multiplied by their assigned scalars, and the length of the current edge is independently multiplied by each of the possible scalars. The likelihood of observing the rates initially estimated given the modified edge lengths is calculated for each potential current scalar, and the potential scalar which maximizes likelihood is assigned to the current edge. This results in each edge of the phylogeny having an associated scalar describing whether the edge has more or less transitions than expected given the length of the edge and estimated rates. We also develop a function that allows for the coloring of edges based on associated scalars. Together, these functions allow for easy quantification and visualization of rate heterogenity in discrete character evolution.
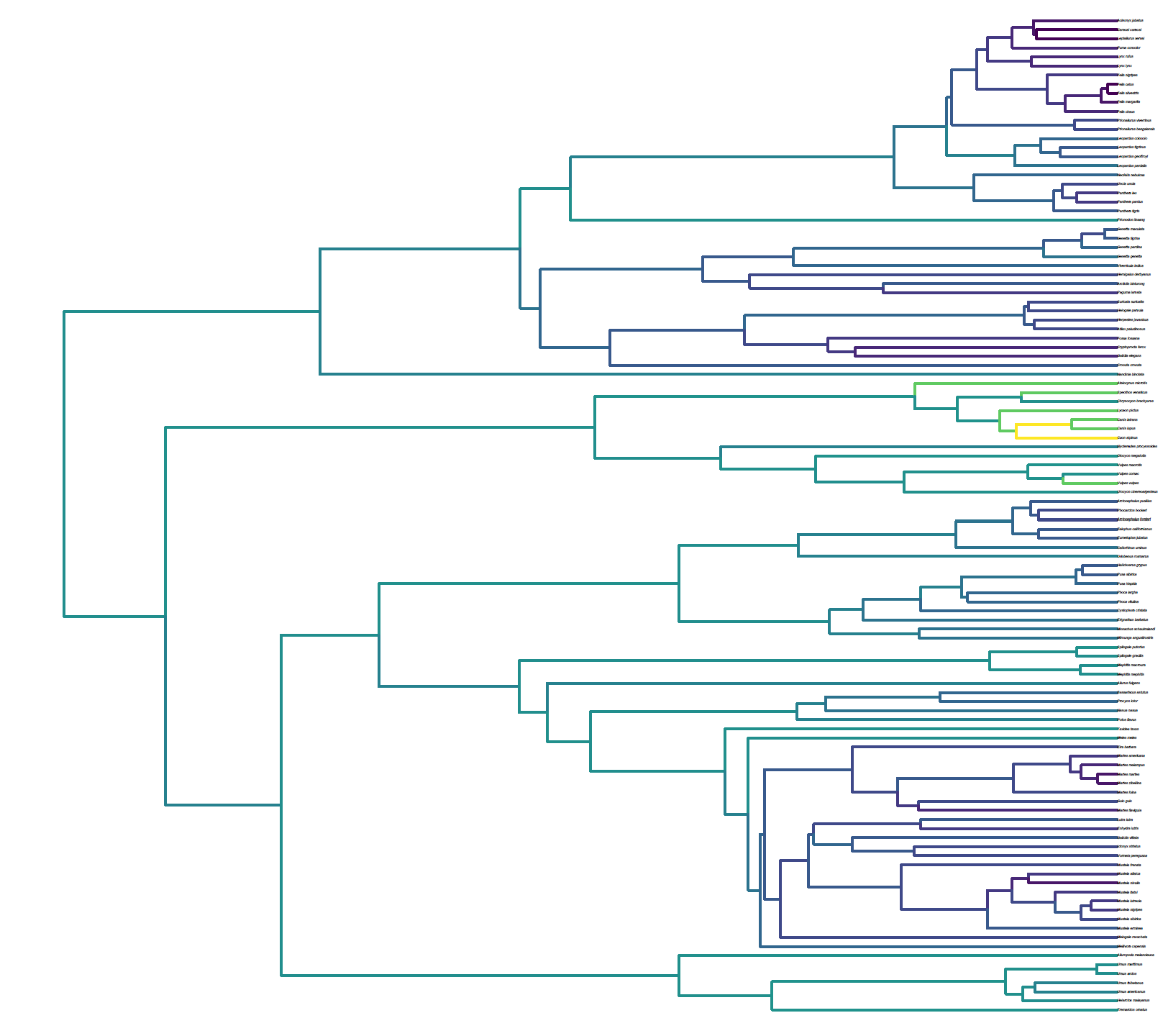
We applied this software to explore variation in rates of karyotypic fusions and fissions in Carnivorans. Our analysis demonstrates that karyotypic evolution occurs rapidly in canids, with this being supported by both observed variation in extent canid karyotypes and historic literature.
Functions for applyihg the rate heterogeneity analytical methods described above can be found here, or in the R package evobiR.
Functions for applyihg the rate heterogeneity analytical methods described above can be found here, or in the R package evobiR.